


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Eichler, R.*; Asai, Masato; Brand, H.*; Chiera, N. M.*; Di Nitto, A.*; Dressler, R.*; D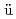 llmann, Ch. E.*; Even, J.*; Fangli, F.*; Goetz, M.*; et al.
llmann, Ch. E.*; Even, J.*; Fangli, F.*; Goetz, M.*; et al.
EPJ Web of Conferences, 131, p.07005_1 - 07005_7, 2016/12
Times Cited Count:3 Percentile:72.98(Chemistry, Inorganic & Nuclear)In recent years gas-phase chemical studies assisted by physical pre-separation allowed for the productions and investigations of fragile single molecular species of superheavy elements. The latest highlight is the formation of very volatile hexacarbonyl compound of element 106, Sg(CO)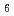 . Following this success, second-generation experiments were performed to measure the first bond dissociation energy between the central metal atom and the surrounding ligand. The method using a tubular decomposition reactor was developed and successfully applied to short-lived Mo(CO), W(CO), and Sg(CO).
. Following this success, second-generation experiments were performed to measure the first bond dissociation energy between the central metal atom and the surrounding ligand. The method using a tubular decomposition reactor was developed and successfully applied to short-lived Mo(CO), W(CO), and Sg(CO).
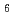 and W(CO)
and W(CO)Usoltsev, I.*; Eichler, R.*; Wang, Y.*; Even, J.*; Yakushev, A.*; Haba, Hiromitsu*; Asai, Masato; Brand, H.*; Di Nitto, A.*; Dllmann, Ch. E.*; et al.
Radiochimica Acta, 104(3), p.141 - 151, 2016/03
Times Cited Count:31 Percentile:94.91(Chemistry, Inorganic & Nuclear)Conditions of the production and decomposition of hexacarbonyl complexes of short-lived Mo and W isotopes were investigated to study thermal stability of the heaviest group 6 hexacarbonyl complex Sg(CO). A tubular flow reactor was tested to decompose the hexacarbonyl complexes and to extract the first bond dissociation energies. A silver was found to be the most appropriate reaction surface to study the decomposition of the group 6 hexacarbonyl. It was found that the surface temperature at which the decomposition occurred was correlated to the first bond dissociation energy of Mo(CO) and W(CO), indicating that the first bond dissociation energy of Sg(CO) could be determined with this technique.
Kurosaki, Yuzuru; Yokoyama, Keiichi; Teranishi, Yoshiaki
Chemical Physics, 308(3), p.325 - 334, 2005/01
Times Cited Count:23 Percentile:60.42(Chemistry, Physical)A total of  1200 trajectories have been integrated for the two dissociation channels of formic acid, HCOOH
1200 trajectories have been integrated for the two dissociation channels of formic acid, HCOOH  H
H O + CO (1) and HCOOH CO + H (2), which occur with 248 and 193 nm photons, using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. It was found that the percentage of the energy distributed to a relative translational mode in reaction 2 is much larger than that in reaction 1. This is mainly due to the difference in the geometry of transition state (TS); the HO geometry in the TS of reaction 1 was predicted to significantly deviate from the equilibrium one, whereas the CO and H geometries in the TS of reaction 2 were found to be more similar to their equilibrium ones. It was also found that the product diatomic molecules, CO and H, are both vibrationally and rotationally excited. The calculated relative population of the vibrationally excited CO for the 248 nm photodissociation was consistent with experiment.
O + CO (1) and HCOOH CO + H (2), which occur with 248 and 193 nm photons, using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. It was found that the percentage of the energy distributed to a relative translational mode in reaction 2 is much larger than that in reaction 1. This is mainly due to the difference in the geometry of transition state (TS); the HO geometry in the TS of reaction 1 was predicted to significantly deviate from the equilibrium one, whereas the CO and H geometries in the TS of reaction 2 were found to be more similar to their equilibrium ones. It was also found that the product diatomic molecules, CO and H, are both vibrationally and rotationally excited. The calculated relative population of the vibrationally excited CO for the 248 nm photodissociation was consistent with experiment.
 CHOCH
CHOCH +CO: Direct ab initio dynamics study
+CO: Direct ab initio dynamics studyKurosaki, Yuzuru; Yokoyama, Keiichi
Journal of Physical Chemistry A, 106(47), p.11415 - 11421, 2002/11
Times Cited Count:36 Percentile:72.97(Chemistry, Physical)A total of 100 trajectories for the photodissociation, CHCHO CH + CO, on the S0 potential surface have been calculated using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. The energy distributions for the relative translational energy, the CO internal energy, and the CH internal energy were calculated to be 28, 20, and 51 %, respectively. It was predicted that the product CO is highly rotationally excited but vibrationally almost not excited; on average, the rotational and vibrational quantum numbers were 68.2 and 0.15, respectively, which qualitatively agrees with the recent observation of Gherman et al. (J. Chem. Phys. 2001, 114, 6128.)
H + Cl CHCl + Cl reaction; Ab initio molecular orbital studyKurosaki, Yuzuru
Journal of Molecular Structure; THEOCHEM, 545(1-3), p.225 - 232, 2001/07
The CASSCF and MRCI calculations with the cc-pVTZ basis set have been carried out for the CH + Cl CHCl + Cl reaction. It has been revealed that the reaction has a small barrier from the CHCl + Cl side at the CASSCF level of theory, but it has no barrier at the MRCI level. Namely, the CHCl + Cl CH + Cl reaction was predicted to be a spontaneous reaction. The result of the MRCI calculation strongly supports the prediction of our previous PMP4(SDTQ) calculation [J. Mol. Struct. (Theochem) 503 (2000) 231].
Yokoyama, Atsushi; Takayanagi, Toshiyuki
Chemical Physics Letters, 307(1-2), p.48 - 54, 1999/00
Times Cited Count:9 Percentile:28.09(Chemistry, Physical)no abstracts in English
Sato, K.*; *; Takayanagi, Toshiyuki; *; Yokoyama, Atsushi
Journal of Chemical Physics, 106(24), p.10123 - 10133, 1997/06
Times Cited Count:31 Percentile:71.5(Chemistry, Physical)no abstracts in English
; Yokoyama, Keiichi;
Chemical Physics Letters, 237, p.106 - 110, 1995/05
Times Cited Count:13 Percentile:45.67(Chemistry, Physical)no abstracts in English
CHClF, CBrFCHBrF, and CBrClFCBrF in a molecular beam; Yokoyama, Keiichi;
Journal of Chemical Physics, 101(12), p.10602 - 10608, 1994/12
Times Cited Count:7 Percentile:30.48(Chemistry, Physical)no abstracts in English
; Yokoyama, Keiichi;
Journal of Chemical Physics, 100(9), p.6487 - 6491, 1994/05
Times Cited Count:30 Percentile:73.11(Chemistry, Physical)no abstracts in English
C(s) and thermochemical properties of gaseous CLi, CLi, and CLiKudo, Hiroshi; Wu, C. H.*
Journal of Nuclear Materials, 201, p.261 - 266, 1993/00
Times Cited Count:15 Percentile:79.48(Materials Science, Multidisciplinary)no abstracts in English
Kudo, Hiroshi
Shitsuryo Bunseki, 41(6), p.317 - 328, 1993/00
no abstracts in English
by Knudsen-effusion mass spectrometryKudo, Hiroshi
Nature, 355(6359), p.432 - 434, 1992/01
Times Cited Count:120 Percentile:96.82(Multidisciplinary Sciences)no abstracts in English
S and LiS molecules in the gas phase over LiS(s,l)Kudo, Hiroshi; Wu, C. H.*
Chem. Express, 5(9), p.633 - 636, 1990/09
no abstracts in English
